本文介紹了將高分離度快速液相色譜法紫外-可見檢測器和四極桿質譜聯用,分析各種中藥(TCM)及中藥制劑的方法,對不同中藥用UV和MS檢測所得到的色譜圖進行了比較,將UV和MS圖譜結合進行目標化合物的鑒定,該方法較傳統控制單一組分的方法更為可靠。
我國擁有長期使用中藥和中藥制劑的歷史,但由于缺乏科學的質量控制標準,中藥一直未能得以飛速發展。如今,對中藥的質量控制僅局限于外觀檢查是遠遠不夠的。隨著新技術的發展,研究人員發現某些中藥中有幾百種化合物,其濃度范圍很寬,而且由于產地和采摘時期不同,以及炮制和加工工藝不同,中草藥的成分也各不相同。因此,對中藥化合物的質量控制極具挑戰性。由于特殊配方的中藥制劑通常是由多種中藥組成,因此只測定中藥制劑中一兩種成分進行質量控制已無法滿足要求。例如用于治療冠心病的中成藥芪參益氣滴丸,就包含黃芪、丹參、三七和降香4種中藥。
本研究以芪參益氣滴丸為樣品,用高分離度快速液相色譜(RRLC)-紫外-可見檢測器和質譜聯用,建立中藥質量控制的新方法。主要目標是建立一種可靠的方法,能夠測定在加熱和混合過程中不變的潛在目標化合物。
實驗內容
實驗儀器
Agilent 1200系列RRLC系統包括:帶真空脫氣機的SL型二元泵、SL型高性能自動進樣器、配置微量流通池(2μl體積,3mm光程)的SL型柱溫箱和二極管陣列檢測器SL;
Agilent 6410四極桿質譜儀,配置ESI離子源;
Agilent 化學工作站B.02.01SR,用于數據采集和分析;
Agilent ZORBAX XDB C18 RRHT色譜柱,3.0×50mm, 1.8μm粒徑。
標準品
丹酚酸B、丹參酮I、丹參酮IIA、隱丹參酮、丹參素、三七皂甙R1和黃芪甲苷均購自中國藥品生物制品檢定所(NICPBP),人參皂甙Rg1、人參皂甙Rb1、原兒茶醛、原兒茶酸、人參皂甙Re和人參皂甙Rc購自Sigma-Aldrich公司(美國)。
溶劑
乙腈購自Fisher公司(美國),水由Milli-Q純水系統制備。
樣品和樣品制備
芪參益氣滴丸、黃芪、丹參、三七的中間提取物,及降香精油,由中國TASLY制藥公司提供。原藥材購自當地中藥店。滴丸、中藥提取物和中藥原料溶于70%甲醇/水溶液中,超聲提取30min,用0.22μm濾膜過濾。
RRLC方法
流動相A:0.1%甲酸水;流動相B:乙腈,含0.1%甲酸;
梯度:0min,10%B;8min,38%B;12min,100%B;持續3min,然后10%B;
流速: 1.0ml/min(被動分流器將進入MS的流速降至0.4ml/min);
柱溫:45℃;檢測波長:203nm;峰寬:0.5s;狹縫寬度:4nm;光譜:190~400nm,2nm步進。
質譜方法
掃描:80~1400(正/負);碰撞誘導解離電壓:70(正/負);干燥氣體:12L/min;
霧化器壓力:50psi;干燥氣溫度:50℃;毛細管電壓:3200V(正/負)。
結果與討論
用安捷倫方法轉換計算器(5989-5130EN)將常規液相色譜方法轉換成高分離度快速液相色譜方法。
丹參三七黃芪和降香提取物的比較
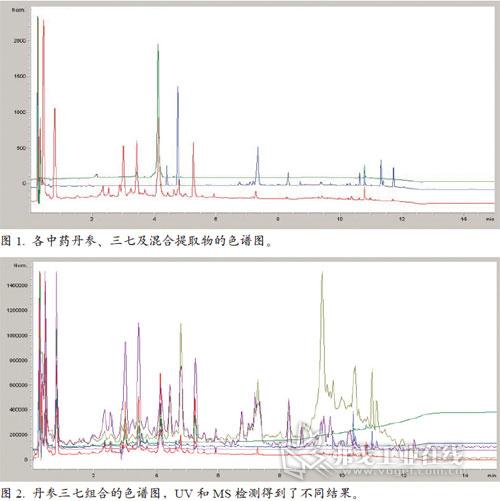
由圖1可見,在203nm檢測波長下丹參和三七的色譜圖與混合提取物有很大區別。混合和加熱后,某些峰消失了,同時出現了一些新的色譜峰。目標化合物篩選是質量控制的重要步驟,可以確定加工過程中這些化合物是否發生了變化。大多數研究者都用203nm進行檢測,因為主要活性化合物是皂甙,在203nm波長下有弱吸收。但203nm處的大峰可能與質量控制無關,那些小峰可能對研究活性成分更重要。因此,需要另一種靈敏度更高的檢測器,提供這些成分的更多信息。
不同檢測器和條件之間的比較
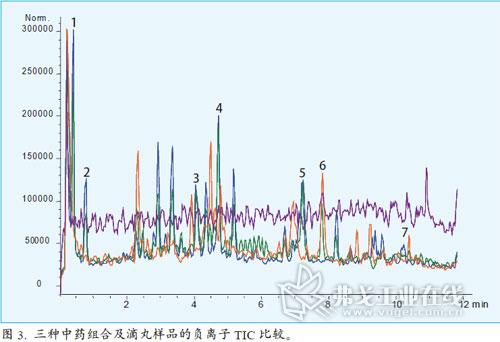
圖2顯示了紫外和MS檢測的不同結果。在MS的總離子色譜圖中有更多的峰,因為某些成分紫外吸收很弱或者沒有吸收。用MS檢測增加了另一維數據,并得到了這些不同化合物的結構信息。負離子模式給出了這些峰更完整的信息,可以對質量控制的目標化合物進行鑒定。
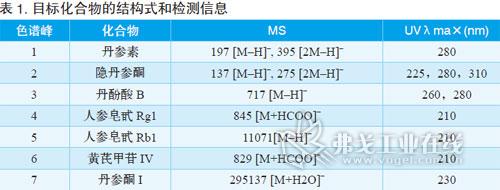
比較芪參益氣滴丸和不同提取物
圖3顯示了丹參、黃芪和降香提取物與芪參益氣滴丸(由這3種提取物和其他填加劑制成)的色譜圖。標記數字的峰就是對這一中藥制劑質量控制篩選到的目標化合物。根據UV和MS數據,研究不同的色譜峰,以確定這些化合物是否發生了改變,鑒別結果如表1所示。
小結
本文介紹的RRLC紫外和MS檢測方法可根據UV和MS檢測器提供的信息跟蹤中藥中幾種成分的質量,因此較目前藥典控制單一組分的質控法更為可靠。由于中藥中的成分可能在炮制和加工過程中發生變化,因此對其的跟蹤非常重要。